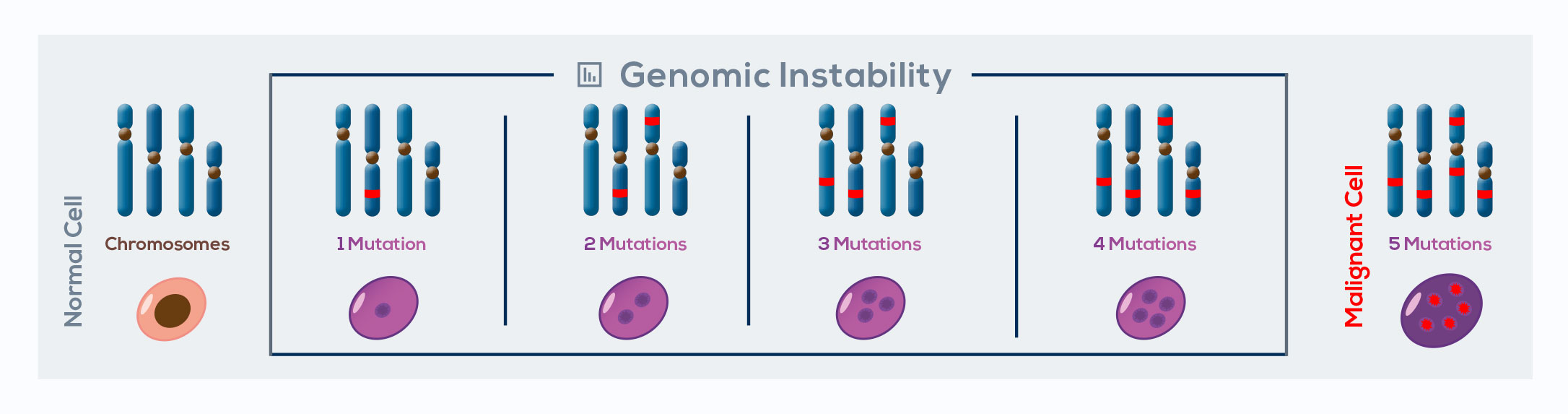
Cancer is a genetic disease that manifests when cells accumulate genomic instability – the principal driver of cancer development – over a period of time and acquire replicative immortality. In combination with other oncogenic changes, telomere shortening can promote chromosomal instability, significantly contributing to genomic rearrangements associated with tumorigenesis. In fact, by protecting chromosome ends, telomeres avoid end-to-end fusion during a normal cell’s life. However, during the M2 crisis, breakage-fusionbridge cycles take place. Sister chromatids lacking protective ends originated by DNA replication fuse together. This phenomenon gives rise to the formation of a bridge of DNA that interferes with the correct segregation of sister chromatids during cell division. When the bridge breaks, uneven derivative chromosomes form, and in the daughter cell a second fusion occurs, developing further genomic instability.
In the majority of cells, high genomic instability leads to cell death. However, in rare cells the activation of telomerase enables escaping from telomere crises. Breakage-fusion-bridge cycles are interrupted by telomerase-mediated chromosome end healing. This leads to the generation of transformed cells with a heavily rearranged, stabilized genome enriched with potentially tumorigenic mutations. Available data link telomere dysfunction to nearly all cancer-related genome alterations:
gain and loss of chromosome (aneuploidy), translocations, gene loss, regional amplifications, whole genome reduplication (tetraploidy), chromothripsis (tens to hundreds of genomic rearrangements in one or few segments of DNA), and kataegis (a specific hypermutated pattern often accompanying chromothripsis). To produce many of these rearrangements and to wreak havoc on the genome, it is sufficient to lose the telomere at individual chromosome ends.