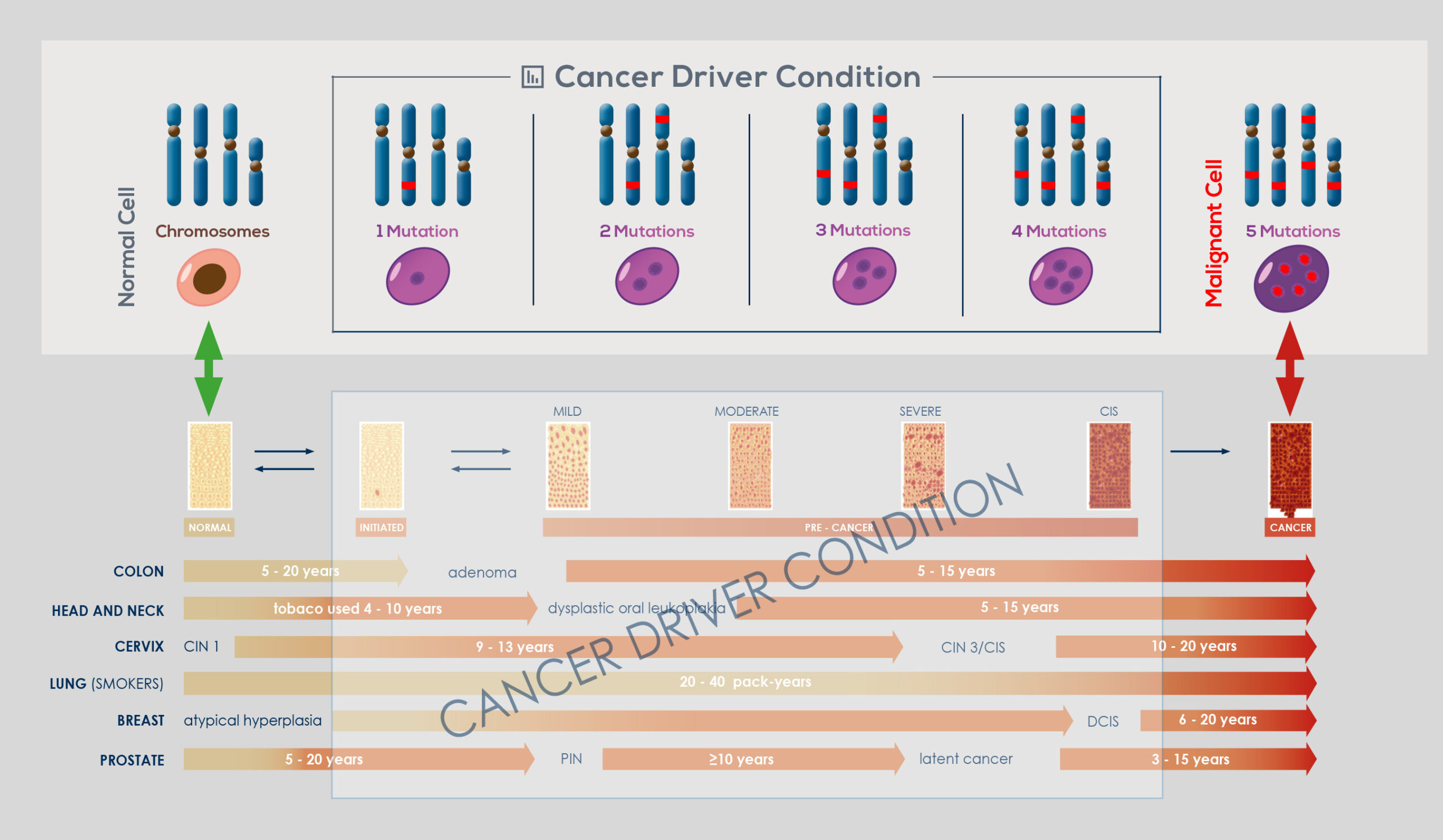
Genomic instability naturally arises in normal cells through accumulation of genetic and epigenetic (that is, those that do not modify gene sequence) changes. This has been shown to occur over a timespan ranging from years to decades, during which the vast majority of cells that acquired it die, and probably are cleared away by the immune system. However, if an even minimal fraction of cells that have acquired genomic instability are not cleared away, it can eventually give rise to a tumor. Selection pressure toward tumor development can arise from phenomena such as the alteration of the mechanisms that control telomere length or responsible for the DNA damage response (DDR) – as demonstrated by the complex mutational signature of smokers’ lung cancer genome or of malignant melanoma, that is cancers that are associated with the exposure to DNAdamaging agents. Most cancers develop genomic instability at some stage of their progression.
In the end, nearly all solid tumors are genetically unstable; or, sometimes, there is a temporary rise in instability, which is followed by the return to a relatively stable genome. Moreover, genomic instability drives intra-tumor genetic heterogeneity, making no tumor identical to another or composed of genetically identical cells. The link between genomic instability and cancer is so strong that upon cancer diagnosis it is possible to take advantage of genomic instability to effectively treat it.
For example, evidence suggests that tumors with more single point mutations respond better to immune checkpoint therapies. Also, it is possible to target compromised mechanisms that lead to genomic instability to kill cancer cells without affecting healthy cells. Finally, genomic instability analysis could give an explanation for resistance to therapy and allows for identifying resistanceassociated genetic alterations to inform treatment decisions and for monitoring tumor burden in response to therapy.
Several studies have been underlying the mechanisms at the basis of genomic instability. DNA integrity is under constant threat. Damage can arise from both endogenous and exogenous factors, in particular spontaneous DNA damaging events (e.g.: modification of DNA bases, or generation of DNA breaks by reactive oxygen species produced by the normal cell metabolism), exposure to external agents (e.g.: ultraviolet (uv) irradiation, or genotoxic chemicals), and failure in cellular DNA processing and replication. The capability to repair damaged DNA has been shown to be essential to preserving genome stability, and the maintenance of genome integrity by the DNA Damage Response (DDR) is critical to preventing cancer development.
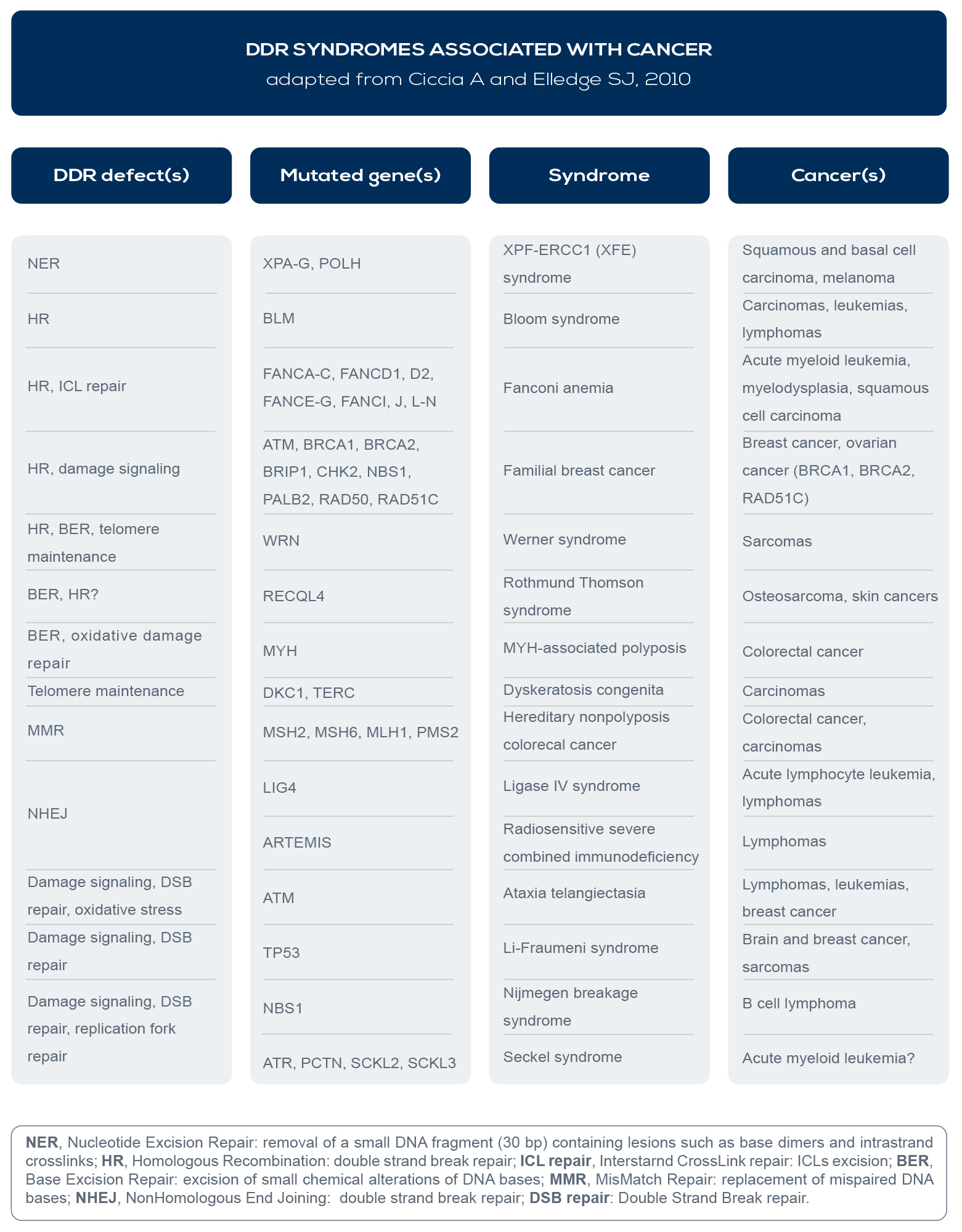
In fact, germline DNA repair gene mutations are associated with cancer predisposition syndromes (e.g.: BReast CAncer gene – BRCA – mutations with breast and ovarian cancer syndromes), suboptimal repair capacity could influence cancer susceptibility, and several DDR syndromes are associated with increased cancer risk (see Table). Inherited or acquired (germline or somatic) mutations leading to the loss of DNA repair efficiency accelerate the accumulation rate of mutation by 100- 1,000 times, and selective pressure favors cancer driver mutations (the socalled “mutator phenotype”).
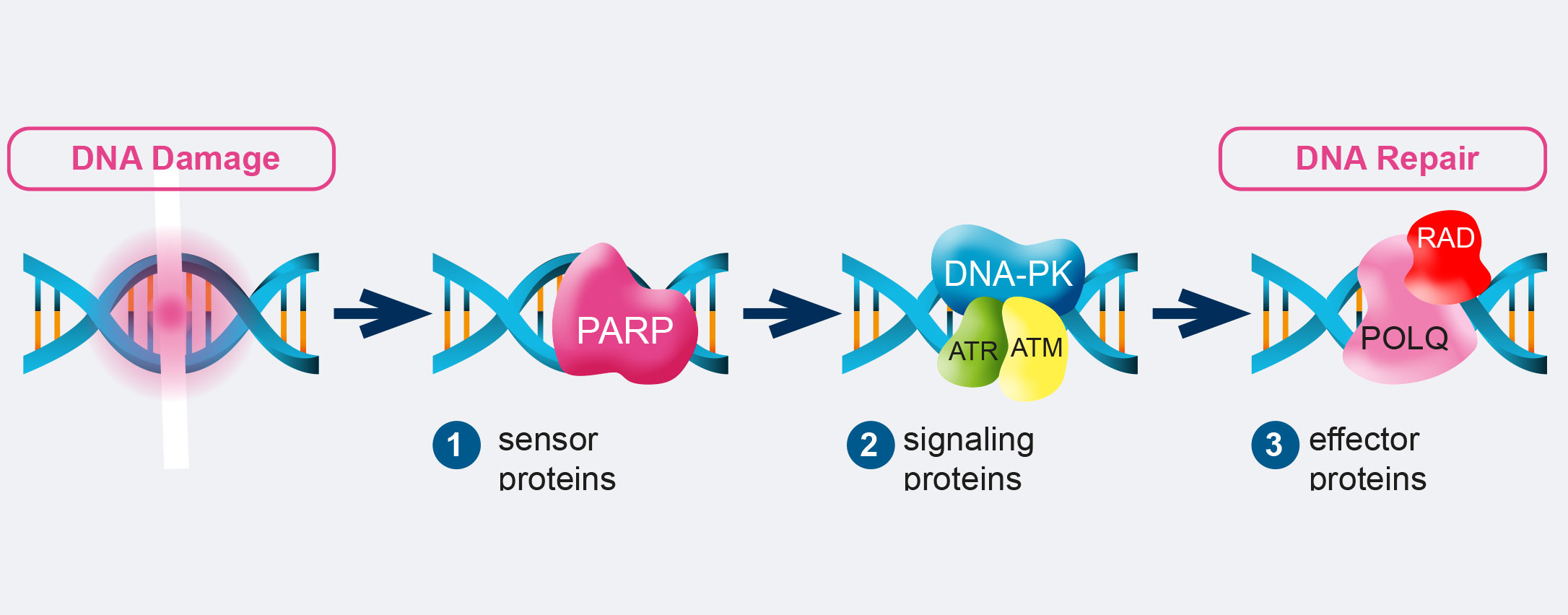
The DDR consist of DNA damage sensors, signal transducers, and protein effectors. The DNA damage sensor machinery can induce apoptosis to eliminate cells that are heavily damaged, or promote DNA damage tolerance. What is more, DDR can activate checkpoints and arrest cell cycle progression to enable DNA repair, promoting genomic stability. Among the principal signal transducers are ATM (Ataxia-Telangiectasia Mutated) and ATR (Ataxia Telangiectasia and Rad3- related protein) kinases. They initiate events that regulate DNA metabolism, cell cycle, and cell fate. Instead, among the enzymatic activities directly involved in DNA repair are nucleases, ligases, polymerases, helicases, topoisomerases, recombinases, glycosylases, kinases, and phosphatases.
For example, RAD50, BRCA1 and 2, and Fanconi Anemia (FA) genes are all involved in Homologous Recombination (HR), the DNA repair pathway that can fix double strand breaks (DSBs), the most lethal form of DNA damage. Mutations in these factors are associated with cancer. For example, ATM is involved in ataxia telangiectasia, a disease which increases lymphoma incidence by roughly 1000-fold. BRCA1 and 2 mutations are linked to breast, ovarian, prostate, pancreatic, and colon cancer. And FA genes are responsible for Fanconi anemia, which is associated with increased breast, ovarian, and oral cancer risk.
The abnormalities that can arise as a consequence of the failure to repair damaged DNA can be grouped into two distinct forms: Somatic Point Mutations (SPMs) and Somatic Copy Number Alterations(SCNAs). SPMs consist of subtle sequence changes. Meanwhile, SCNAs involve gene amplification, loss or gain of whole chromosomes, or chromosome translocations which can result in gain or loss of chromosomal material or in the generation of new gene products. For example, oncogene amplification occurs in a subset of many late-stage cancers, clearly associates with tumor progression, has prognostic significance, provides targets for therapeutics, and represents a common way for cultured cells to acquire resistance to chemotherapy. Both SPMs and SCNAs are linked to cancer. In general, evidence supports a greater role for SCNAs in developing and maintaining cancer cell population diversity, while uncommon SPMs cause dramatic phenotypes. However, the simple presence of genetic alterations, even when frequent, is not a marker of genetic instability. In fact, by definition, instability is a matter of rate. That is why the simple analysis of SPMs or SCNAs cannot be used to determine genetic instability. Unfortunately, most studies on cancer genomic instability analyze somatic mutations at a single time point, but to accurately estimate somatic mutation rates in an individual, samples from at least two time points are needed. In fact, a single high mutation frequency can be a random error causing incorrect evaluations. Instead, a somatic mutation frequency growth trend, manifested over a prolonged period, is indicative of genomic instability.