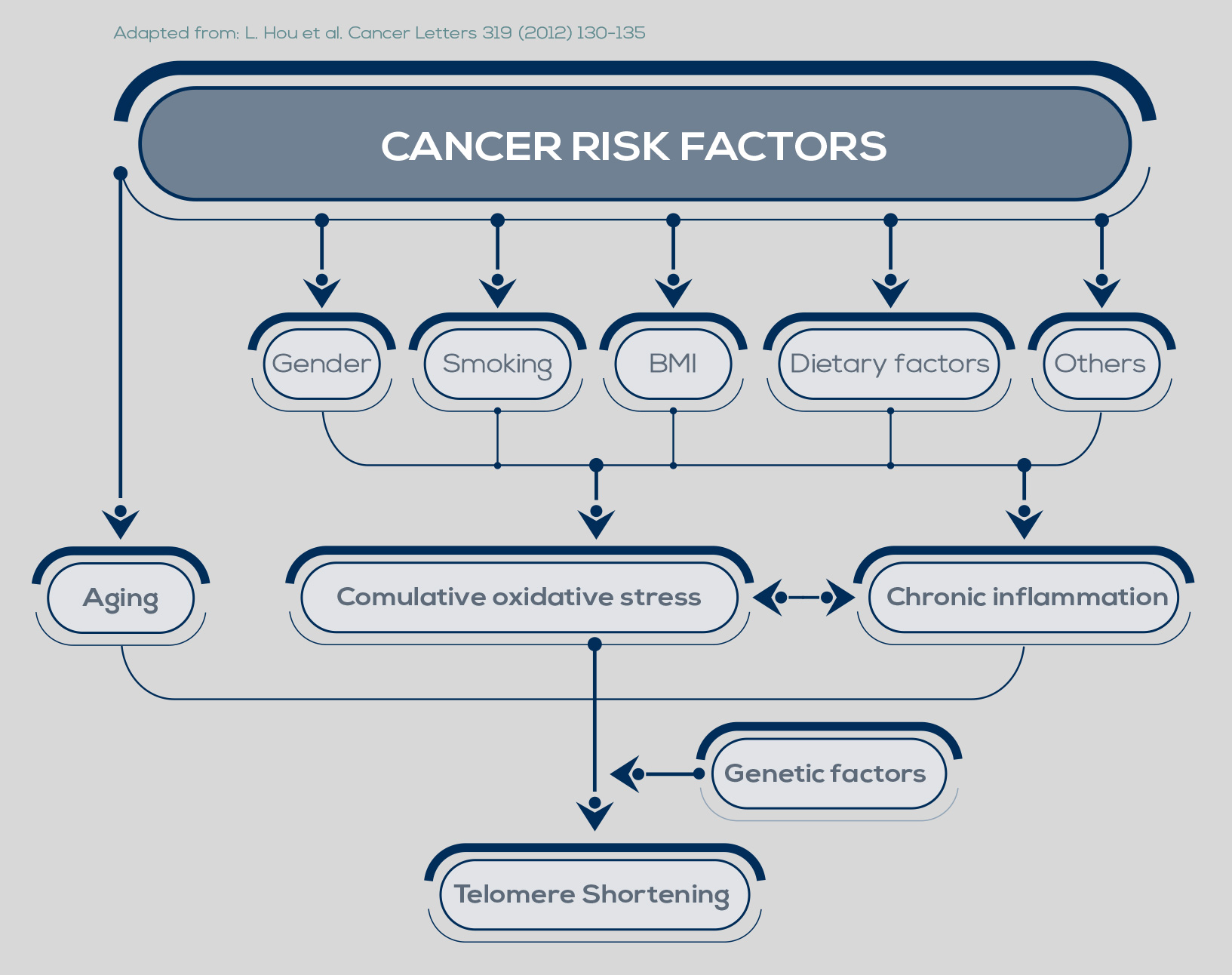
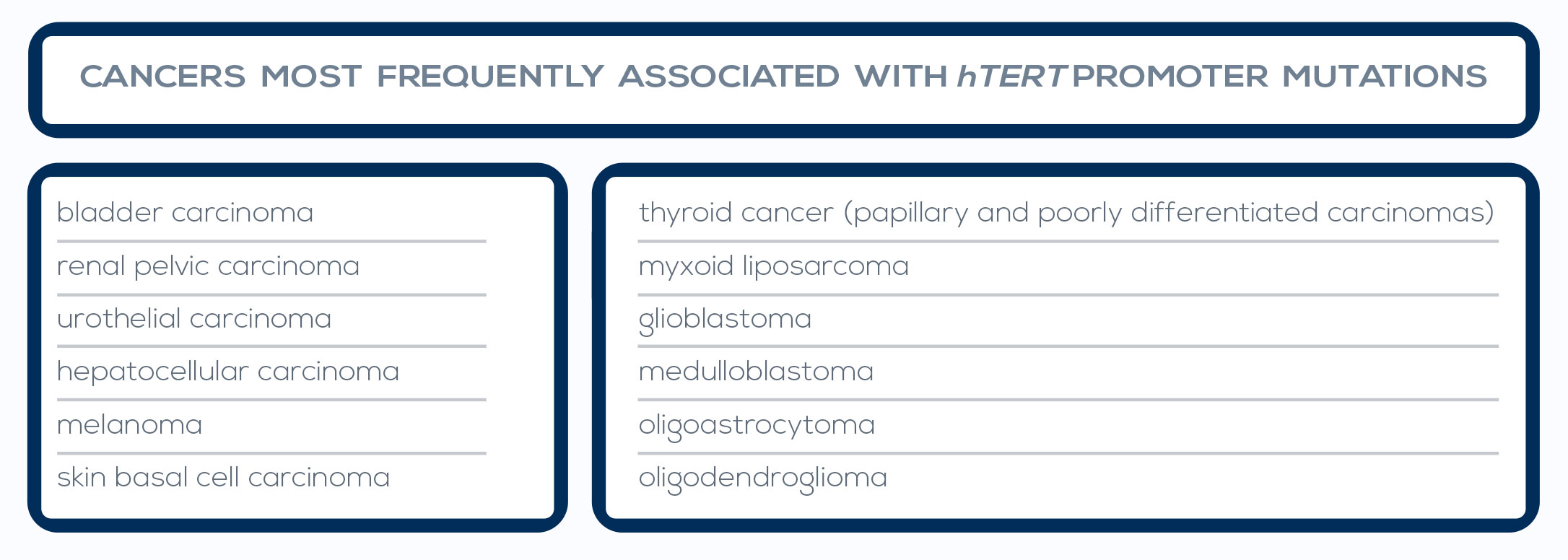
Telomere shortening is a natural phenomenon that occurs with aging. Its cause lies in the mechanism of DNA replication. In fact, one of the two strands of the DNA double helix cannot be copied all the way to its very end, leading to the loss of part of the protective cap of the chromosome at every cell cycle (50- 200 bp, depending on the tissue). After 50 to 60 cell divisions, telomeres are short enough to uncap the chromosome end, and their shortening can promote genome instability.
Inflammation and oxidative stress, two major pathways of carcinogenesis, accelerate telomere shortening. Moreover, the progressive reduction of telomere length is particularly dangerous when combined with changes such as loss of the function of p53, a protein that inhibits cell proliferation and induces senescence, or of other cell-cycle regulators.
In fact, during the period of replicative senescence (or mortality stage 1, M1) that follows the uncapping of one or two shortened telomeres, cellular proliferation is usually inhibited. However, oncogenic changes can promote an M1 bypass, thus extending the cell division period during which telomeres can further shorten. During this new dysfunctional state (the M2 crisis), chromosome end-toend fusions can occur. The cell death triggered by this situation protects from cancer development. However, between 1 in 100,000 and 1 in 10 million cells can develop mechanisms to maintain its very short telomeres, bypassing the M2 crisis and becoming immortalized.