More than 20% of all cancers are initiated or exacerbated by inflammation. For example, hepatocellular carcinoma, gastric cancer, and colon cancer have all been associated with chronic inflammation. Gastritis, the inflammation of the lining of the stomach, correlates with gastric cancer, whereas colitis, the inflammation of the colon, associates with colon cancer. Also, non-infectious causes of chronic inflammation, such as hormonal changes, cigarette smoke and other toxicants (for example adhesives, air fresheners, cleaning products, and glue) increase cancer risk. It is estimated that almost 20% of smokers with inflammation of their bronchial tubes’ mucous membranes (a bronchitis) can develop cancer. Moreover, specific variants of inflammatory genes such as tumor necrosis factor (TNF) and IL-1 and mutations in genes playing important roles in innate immunity have been associated with cancer. For example, the aberrant activation of NF-kB or STAT3, which are involved in inflammation control, is found in over 50% of all cancer; this activation renders both premalignant and fully transformed cells resistant to apoptosis, and accelerates their rate of proliferation, increasing tumor growth. On the other hand, non-steroidal anti-inflammatory drug (NSAIDs) use has been associated with a 30-60% reduction of colon cancer incidence. In general, pro-inflammatory cytokines have been implicated in inflammationassociated tumors.
This association is supported by epidemiological, genetic, and pharmacological studies. Inflammation can contribute to cancer initiation through mutations in genes involved in cell division, survival, senescence, or genomic instability (e.g. genes involved in error correction after DNA replication), or promoting chromosome instability and DNA double strand breaks. It can also promote cancer development through cellular and extracellular signals making cells resistant to growth-inhibitory signals, apoptosis, or antitumor immunity. It acts both via extrinsic pathways (that is, via chronic conditions associated with smoldering, non-resolving inflammation) and intrinsic pathways (that is, genetic events that orchestrate the generation of cancer-related inflammation), playing a crucial role in several aspects of tumor development, including cellular transformation, cell survival, cell proliferation, invasion, metastasis formation, and angiogenesis. A key event in inflammation-related carcinogenesis is the production of reactive oxygen and nitrogen species (ROS and RNS). These highly reactive molecules damage DNA promoting cancer development (e.g. cholangiocarcinoma related to liver fluke infection), can induce irreversible and irreparable protein changes, and can contribute to the generation of reactive lipids that can further interact with and damage DNA and proteins.
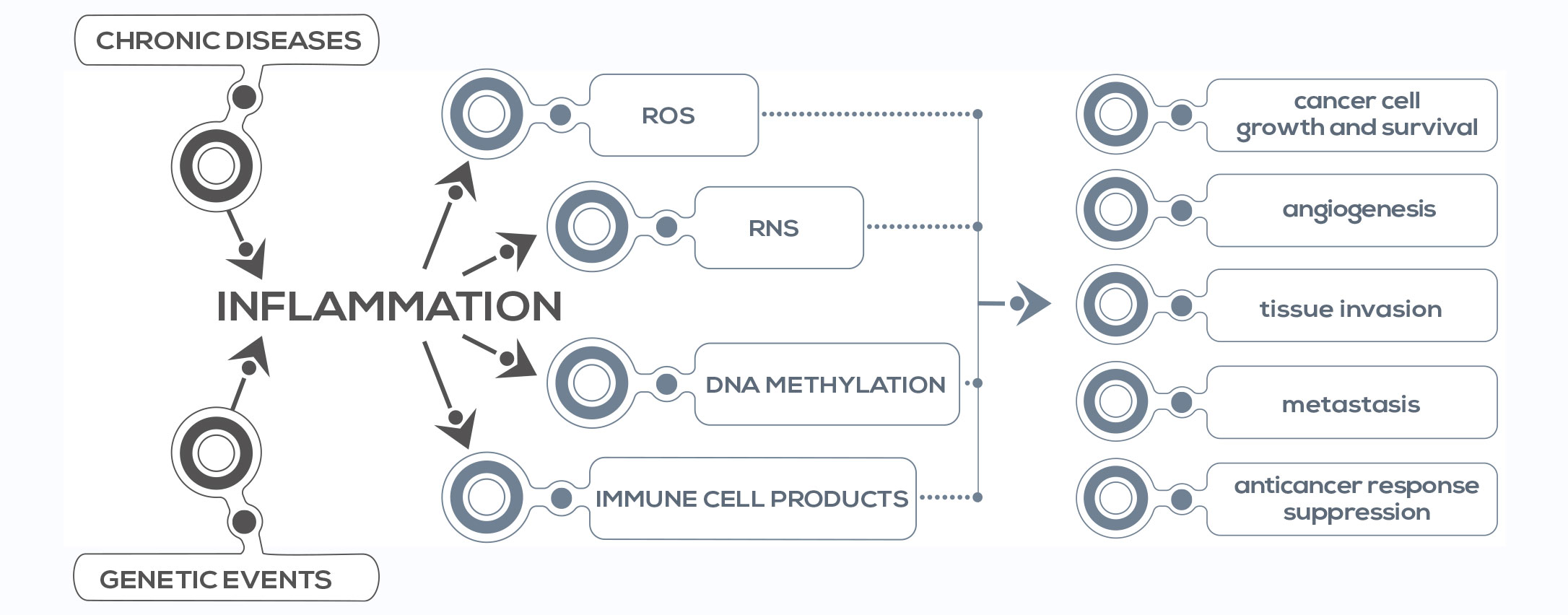
All these lesions result in molecule disfunction, damaging tissues and activating stem cells for tissue regeneration. Moreover, ROS and RNS damage stem cells too, resulting in mutation accumulation and, THE ROLE OF INFLAMMATION IN CANCER eventually, cancer stem cell generation. Damage to cancer stem cells’ DNA is associated with more aggressive cancer. Another pivotal event is DNA methylation – the addition of a chemical group (methyl) to specific DNA sequences. ROS and RNS induce global DNA hypomethylation, resulting in genomic instability, and inflammatory cytokines such as IL-6 affect the expression of the gene encoding for DNA methyltransferase 1 (DNMT1), enhancing, for example, the methylation of tumor suppressor genes – thus downregulating their expression, an event that can represent the first step of carcinogenesis. Finally, chronic inflammation is associated with immune suppression. Ongoing inflammatory responses can suppress anticancer immune responses. In fact, tumor-associated immune cells can produce immunosuppressive cytokines such as IL-10 and transforming growth factor (TGF-β) and other molecules that negatively influence immune responses against cancer cells.
